
ATTR-CM overview
 Fact-checked by
Fact-checked by Transthyretin amyloid cardiomyopathy (ATTR-CM) is a progressive condition that can be caused by genetic mutations or aging, and is characterized by accumulating heart damage that ultimately leads to symptoms of heart failure.
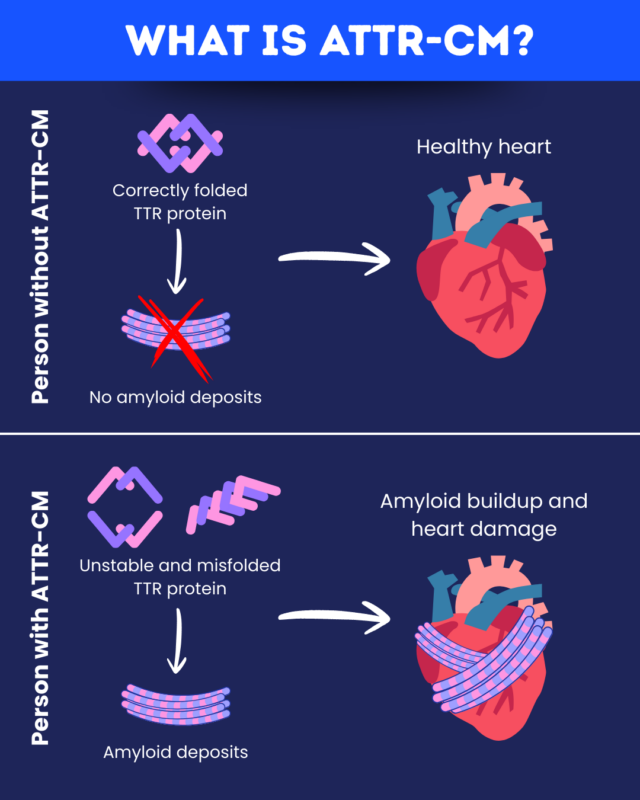
In ATTR-CM, the protein transthyretin (TTR) misfolds and forms clumps called amyloid deposits that build up in the heart and other tissues.
Because its symptoms can mimic other types of heart disease, ATTR-CM is difficult to diagnose, but early recognition is critical. It allows for the prompt initiation of disease-modifying therapies to slow disease progression, as well as supportive care to improve life quality.
What is ATTR-CM?
ATTR-CM is a type of amyloidosis, a group of diseases in which amyloid deposits accumulate in the body’s tissues. It is one of the most common types of cardiac amyloidosis, where these protein clumps accumulate in the heart.
Because the amyloid deposits in ATTR-CM contain a misfolded version of TTR, it is considered a type of transthyretin amyloidosis. When TTR deposits mainly affect the nerves, a related disease called hereditary transthyretin amyloidosis with polyneuropathy (hATTR-PN) may be present instead. Many people with these diseases experience a combination of heart and nerve issues.
Types of ATTR-CM
The two primary ATTR-CM types are:
- hereditary ATTR-CM, caused by genetic mutations in the TTR gene that encodes TTR
- wild-type ATTR-CM, which typically develops with age
In hereditary ATTR-CM, disease-causing mutations are passed down in families. The age of onset can vary, but symptoms often begin in the 50s or 60s. This form is more likely to be accompanied by some degree of nerve damage.
Wild-type ATTR-CM isn’t related to genetic mutations, can’t be inherited, and primarily affects older men. It is typically diagnosed after age 70. Symptoms related directly to nerve damage are not as prominent, but may still occur.
Causes of ATTR-CM
The TTR protein usually exists as a stable four-part structure. In ATTR-CM, TTR’s structure becomes unstable, causing the protein to dissociate into individual subunits that misfold and form amyloid deposits.
There are two main underlying ATTR-CM causes that lead to TTR instability.
- In hereditary ATTR-CM, mutations in the TTR gene lead to the production of an unstable TTR protein. Different TTR mutations can lead to ATTR-CM, hATTR-PN, or a mixed form of transthyretin amyloidosis. Notably, not everyone who inherits a mutation will develop symptoms.
- In wild-type ATTR-CM, cells initially produce a normal TTR protein that later becomes abnormal and misfolds, likely due to age-related processes that make it less stable.
Symptoms of ATTR-CM
Cardiac ATTR-CM symptoms may include:
- shortness of breath
- fatigue
- swelling in the feet and ankles
- abnormal or fast heart rate
- difficulty exercising
- chest pain
- coughing or difficulty breathing when lying down
- abdominal bloating
- confusion or trouble thinking
These are typical symptoms of heart failure and may be similar to those of other heart diseases.
While the heart is the main organ affected in ATTR-CM, nervous system problems are not uncommon and may include:
- pain, tingling, or other abnormal sensations in the hands or feet
- carpal tunnel syndrome, characterized by pain in the hands or forearms
- spinal stenosis, characterized by pain in the lower back
- issues with involuntary body functions, such as gastrointestinal issues, low blood pressure when standing up, erectile dysfunction, urinary problems, or abnormal sweating
These may be among the earliest signs of ATTR-CM, appearing even years before overt heart issues. Tendon tears, eye issues, or kidney problems are also possible.
ATTR-CM diagnosis
A patient is usually evaluated for ATTR-CM if they have red flag symptoms that raise suspicion of the disease, especially in the presence of risk factors such as older age, male sex, or a family history of ATTR-CM.
A formal ATTR-CM diagnosis may then involve a combination of the following tests:
- echocardiography and electrocardiography to monitor heart structure and function
- blood and urine tests to exclude other causes of amyloidosis
- scintigraphy scans, which can identify amyloid deposits in the heart
- tissue biopsy or cardiac MRI to confirm the presence of amyloid, particularly if other tests are not conclusive
After the diagnosis of ATTR-CM is established, genetic testing can be used to determine whether the disease is hereditary or wild-type, which may have implications for treatment and monitoring.
ATTR-CM treatment
ATTR-CM treatment is a multidisciplinary effort that involves disease-modifying and supportive therapies.
Disease-modifying therapies aim to reduce amyloid accumulation in order to slow disease progression in both hereditary and wild-type ATTR-CM. These include:
- Gene silencing therapies, which suppress TTR production. Amvuttra (vutrisiran) is the only one approved in the U.S. for ATTR-CM.
- TTR stabilizers, which work by stabilizing the protein’s structure to prevent it from misfolding. The approved therapies are Attruby (acoramidis) and Vyndamax (tafamidis).
These therapies cannot reverse existing organ damage or clear amyloid deposits that have already formed, so early initiation is crucial.
Supportive therapies, including medications and lifestyle changes, can also help ease symptoms and improve life quality.
How common is ATTR-CM?
Accurate data on the true number of people living with ATTR-CM are limited, but it is generally believed that the disease remains an underdiagnosed cause of heart failure.
In the U.S., it has been estimated that approximately 6.1 per million people have ATTR-CM, with higher rates in other regions, including Nordic countries and Japan. In Sweden, approximately 50 people per million are estimated to have ATTR-CM.
The disease is more prevalent in high-risk groups, including older men and people with heart failure.
It is generally reported that wild-type ATTR-CM is the more common form of the disease, although this may vary by geographical region. Different reports have found that anywhere from 5% to 60% of people with ATTR-CM have the hereditary form.
ATTR-CM prognosis
As a progressive disease, ATTR-CM can ultimately cause serious heart damage, and death from cardiac complications is common.
- In wild-type ATTR-CM, the median survival after diagnosis is around 3-5 years.
- The median life expectancy in hereditary ATTR-CM varies depending on the specific TTR mutation, ranging from around 2.5 years with the aggressive Val122Ile mutation to closer to six years with milder variants.
Several factors, including coexisting conditions, disease severity at diagnosis, and access to treatment, may affect an individual’s prognosis. With the recent emergence of disease-modifying therapies that slow disease progression, the life expectancy for ATTR-CM is improving.
There is no cure for ATTR-CM, but careful monitoring and early treatment can help minimize its impact on daily life. Regular follow-up with a multidisciplinary care team is vital for tracking disease progression and making necessary adjustments to the care plan.
Amyloidosis News Today is strictly a news and information website about the disease. It does not provide medical advice, diagnosis, or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.


